Military Parents Keep the Faith With Son’s SMA Diagnosis

Sgt. 1st Class Kanaan Merriken recites the oath of enlistment as his wife Kari and son Caleb look on. Merriken and his wife Keri have focused on raising public awareness of SMA while providing the care their son needs. 75th Ranger Regiment photo by Tracy A. Bailey
Fort BENNING, GA — Kanaan and Kari Merriken had never heard of spinal muscular atrophy until their son was diagnosed with the disease in 2010. At the time, Kanaan was a staff sergeant with the 75th Ranger Regiment at Fort Benning. Since then, Kanaan, now a first sergeant, and Kari have focused on raising public awareness of SMA while providing the care their son needs.
“No parent ever wants to be told that their child has SMA,” said Mary Schroth, MD, chief medical officer of Cure SMA. “Not that long ago, parents of newly diagnosed SMA babies were told to take them home and love them and that their lives will be short—usually less than two years.”
The Merrikens were up to the challenge. Kanaan had already survived extensive wounds including a damaged carotid artery and brain injury from an improvised explosive device attack in Baghdad in 2003. Not expected to live, he was Medevaced to Landstuhl Regional Medical Center in Germany and medically retired. To the surprise of his physicians, he recovered, then returned to the Army and the Rangers in 2005—and competed in the Best Ranger Competition in 2011. Throughout, Kari has relied on her deep faith.
First described nearly 130 years ago, spinal muscular atrophy (SMA) remains the leading genetic cause of death for infants. That grim fact and the classic categorization of the disease may soon fade into history as new therapies and increased awareness of the genetic underpinnings of SMA revolutionize treatment.
In nearly all cases of SMA, a deletion of or mutation in both copies of the survival motor neuron 1 (SMN1) gene in chromosome 5 prevents production of the SMN protein essential to maintaining healthy motor neurons, nerve cells in the brain stem and spinal cord that send signals to the muscles in the tongue, throat, face, chest, legs and arms. Without the protein, the neurons die. Without signals from the neurons, the muscles they control weaken and atrophy. As the muscles weaken, activities such as breathing, swallowing, speaking, and walking become more difficult.
About one in 40 individuals carry the recessive mutation primarily responsible for SMA, which affects between one in 6,000 and one in 10,000 live births. Mutations in other genes, including IGHMBP2, VAPB, DYNC1H1, BICD2, and UBA1 can cause rarer forms of SMA and SMA-like diseases through different mechanisms.
Classic SMA
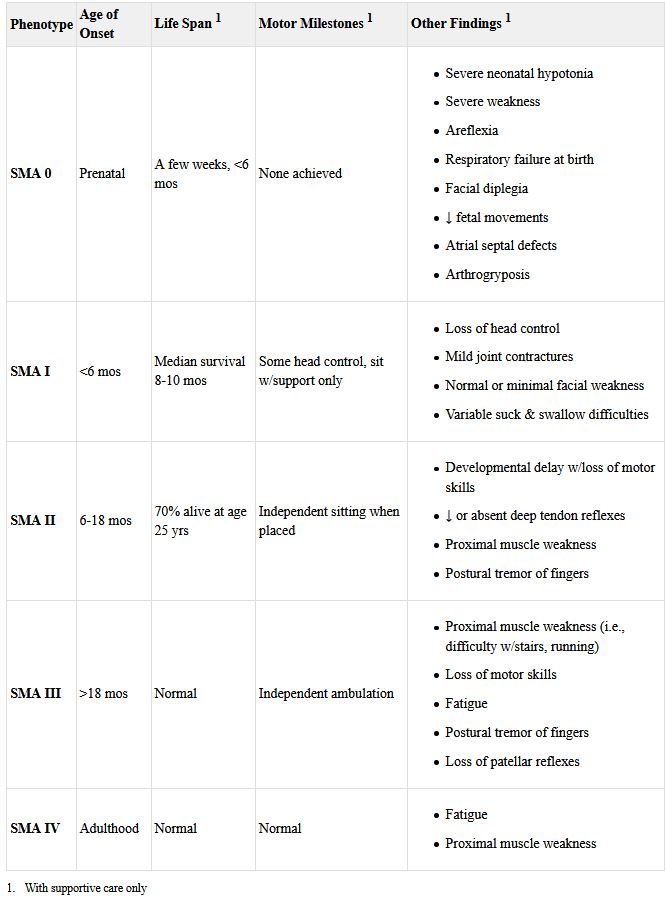
Spectrum of SMA Phenotypes at Presentation
SMA has been traditionally been divided into four main types. Type 1 manifests before six months of age. Also called infantile-onset SMA or Werdnig-Hoffman disease, neonates with the disease appear to be “floppy infants” with reduced movement, lack of tendon reflexes, muscle contractures, and difficulty swallowing and feeding. Without treatment, these children never sit on their own and typically die before age two of respiratory failure.
“It’s not as rare as you might think,” Kanaan Merriken told The Bayonet, Fort Benning’s newspaper, shortly after his son’s diagnosis. “My co-worker’s son has spinal muscular atrophy type 1. He can’t move his legs. He can’t roll over. They have to have him on oxygen.”
The Merriken’s son was diagnosed with type 2 SMA (Dubowitz disease), which develops between six and two years of age. At the time of his diagnosis, there was no way to stop disease progression. These children typically manage to sit without support, but do not stand or walk without assistance. In time, they may lose the ability to sit unaided. Curvature of the spine is common. They often have respiratory challenges and reduced life expectancy without treatment.
Type 3 SMA or Kugelberg-Welander disease develops after 18 to 24 months, sometimes first appearing in older children or adolescents. Individuals with this type can generally walk independently, though they may have trouble running or climbing stairs. Scoliosis and muscle contractures are common. Life expectancy is not reduced, but affected individuals often experience increased instability and reduced mobility that may require a wheelchair as they age.
Individuals with type 4 SMA develop symptoms as adults, often after age 35. They have the mildest course, with the disease primarily affecting the leg muscles. Fatigue is common as is mild tremor or occasional muscle cramping.
Some schemas include a Type 0, in which infants suffer prenatal onset and live just a short time.
A New View
For years, the Merrikens and other families affected by SMA held 5K runs and other programs to raise money to fund development of treatments that would change the trajectory of the disease—and they succeeded.
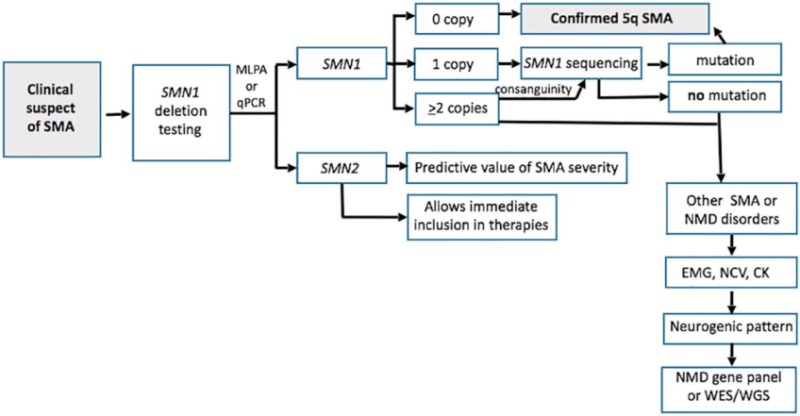
Click To Enlarge: Diagnostic algorithm for SMA
Researchers discovered a crucial factor in the speed and severity of development of SMA: the number of copies of a second gene that can also produce the SMN protein. All patients with SMA have at least one copy of this back-up gene, SMN2, which is 99% similar to the SMN1 gene. Typically, however, SMN2 produces just 10% of the amount of full-length SMN protein that a functional copy of SMN1 would.
Generally, infants with type 1 SMA have two SMN2 genes, while those with type 2 usually have three copies and individuals with type 3 have three or four copies. Patients with type 4 SMA have from three to eight copies of the back-up gene. Because these genes produce some of the critical protein, more copies often mean later development of the symptoms.
SMN-enhancing therapies that modify SMN2 so that the existing copies produce more full-length proteins or insert functional SMN1 genes have been shown to fundamentally change the progression of the disease, making typing based on timing of manifestation of symptoms or limitations arguably moot for treated patients.
As an example, one study showed that enhancing SMN2 function in presymptomatic infants with two copies of the gene who were treated before six weeks of age enabled 100% of them to sit, 88% to walk, and 77% to walk independently. None needed permanent ventilation and all lived.1 In the traditional classification of SMA, those patients would primarily have been classified as type 1, in which none of those milestones would have ever been achieved. Studies with other approved agents have also produced improvements that call into question the usual typing.
As a result of this shift, some in the field suggest that classification of SMA phenotypes should be updated to include the number of SMN2 copies, the age at treatment initiation, and age of symptom onset.2
With the emergence of disease-modifying therapies, many experts have urged states to incorporate SMA into newborn screening tests. So far, 31 states covering 65% of U.S. babies have adopted or are piloting the test.
An expert consensus-based SMA treatment algorithm recommends using the results of newborn screening to enable initiation of the life-changing treatment in infants with two to four copies of SMN2 immediately. 3 In infants with type 1 SMA, an estimated 90% of motor neurons die within the first six months of life without treatment and they do not regenerate, so time is of the essence.
The panel concluded that infants born with just one copy of SMN2 were likely to be symptomatic at birth and the decision whether to treat should be left to the physician and family based on the child’s disease state.,4
Children who were born before the new treatments were available and patients not treated in the first few months of life may also benefit from therapies approved for these older groups, with studies demonstrating positive results ranging from stabilization of symptoms to significantly enhanced motor skills and even attainment of normal motor milestones.5
- Finkel R, Chiriboga C, Vajsar J, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017-3026.
- Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy—new phenotypes, new challenges, new implications for care. J Neuromusc Dis. 2020;7(1):1-13. doi:10.3233/JND-190424
- Glascock J, Sampson J, Haiet-Phillips A, et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromusc Dis. 2018;5(2):145-158. doi:10.3233/JND-180304
- Glascock J, Sampson J, Connolly AM, et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromusc Dis. 2020;7(2):97-100. doi:10.3233/JND-190468
- Mercuri E, Barisic N, Boespflug-Tanguy O, et al. SUNFISH Part 2: Efficacy and safety of risdiplam (RG7916) in patients with Type 2 or non-ambulant Type 3 spinal muscular atrophy (SMA) Presented at the American Academy of Neurology Conference 2020. Neurology. 2020;94:1260.